Appropriate Acceleration in Clinical Development: 2022 Health Trends
2022 Health Trends
Twelve years. That’s a number we’re all incredibly familiar with. It’s how long it typically takes to bring a new drug to market.
The global pandemic allowed no such timeline. Even as COVID-19 interrupted important studies in many countries, clinical teams continued to drive speed in research and development that we’ve never seen before, engaging patients around the world and creating unbelievable volumes of data. Along the way, we learned what was possible in a crisis —or, part of what was possible. For every COVID-19 trial working at massive speed and scale, there were many other trials for chronic and acute diseases that realized their own forms of innovation to engage sites and patients in a radically changed world.
These experiences changed the expectations of sponsors, sites, advocates, patients and regulators alike. And they will forever change how innovative new therapies are discovered and developed.
In 2022, clinical development will be focused on appropriate acceleration, driven by innovative study design, real world data engines, flexible decentralized engagement and dynamic contracting.
Appropriate Acceleration in Clinical Development
The critical piece bringing all of those together is patient-powered trial design that understands burden, hears and integrates patient voices, and consistently shares data and insight back with participants. That aspect of appropriate acceleration is both so significant and so interconnected with commercial strategies that it has its own trend in this book. For now, we'll focus on the mechanics of the Appropriate Acceleration model.
Innovative Study Design
To optimize speed and improve the likelihood of completing successful, valuable research, sponsors are increasingly focused on two specific kinds of design: synthetic data and platform trials.
1. Synthetic Data
While randomized clinical trials remain the gold standard for evaluation of experimental therapeutics, they present challenges for both patients and sponsors. Reliance on placebo control arms can create challenges in both recruitment and retention. Some patients may be uninterested in taking a chance of receiving a placebo or have already rejected the standard of care. In the trial itself, patients may drop out if they suspect they aren’t receiving any level of treatment.
Synthetic data uses information previously collected in place of a control arm. That data may come from historical control data, real world data, or the generation of a companion data set from other sources.
That data creates an external control arm that becomes the comparator, meaning that every patient in the active study will receive the experimental treatment. While external control arms were certainly used before COVID-19, they’ve been thrown in the spotlight as both a moral imperative and a source of speed in 2022.
The ethical advantages of external control arms range from removing selection biases and easing concerns around placebos to ensuring strong real-world comparators within carefully controlled trials. The speed factor is undeniable—and likely why this approach has been brought into that spotlight now—because sponsors need only enroll half the usual number of patients. That gets trials completed more quickly and approved therapeutics to people faster.
Both the US Food and Drug Administration (FDA) and European Medicines Agency (EMA) have been active in working with sponsors using synthetic data. In the years between 2000 and 2019, the FDA approved 45 treatments with an external control arm.2 Twenty European countries expanded the label for a non-small cell lung cancer treatment based on a similar design. NICE has continued to evaluate the use of external data in its submissions and found increasing use over the last two years.
2. Platform Trials
Platform trials may be the great signal of the industry’s commitment to appropriate acceleration. They’ve challenged a series of steps that previously worked sequentially to work simultaneously. Or, at least with important overlap.
Working from one master protocol, platform trials allow multiple treatments to either enter or exit the trial over the course of the study. That gives researchers the ability to adapt based on the response from different cohorts. They can quickly drop treatments that are not performing and declare one or more approaches superior. The interim data may even lead them to add assessments or treatments that might ultimately best reveal impact for a particular population. Moreover, patients may be matched more effectively with the appropriate trial protocol through streamlined screening measures, reducing the burden on both patients and sites.
Platform trials are not new. But they proved their value during the pandemic—not just in the US, but in Europe. A large platform trial in the UK called RECOVERY was the first to prove that dexamethasone significantly reduces deaths among hospitalized COVID-19 patients, and the same platform further showed that the malaria treatment hydroxychloroquine is of little use.3 These results had an immense impact on clinical responses to COVID-19 around the world.
In 2022 and beyond, you’ll see this approach increasingly used in drugs expected to have multiple indications, letting investigators learn about multiple uses at once. They’re also expected to play critical roles for investigational treatments that only have a very limited period of time to recruit, due to resources, patient need or the competitive environment. In those cases, biopharma leaders are increasingly turning to platform trials where the traditional models won’t work.
As more sponsors adopt platform approaches, look for new conversations about data speed, interim evaluation considerations and statistical rules to prompt action.
Real World Data Engines
Biopharma’s approach to integrating real world data into overall development has moved from hesitancy to reliance. Synthetic arms (discussed above) are one use of real world data, but within the new clinical model, real world data is also building engines of discovery that fuel continuous improvement. Although there are many, three particular "engines" stand out as we enter 2022:
1. Right-now understanding
Randomized controlled clinical trials are a snapshot in time. They’re how particular people responded to a particular drug given a particular pathology. Investigators are using real world evidence to respond when one of those variables changes. A clear example is COVID-19. The vaccine clinical trials were built around the original variant of the disease. As the coronavirus spread and mutated, new variants emerged that simply didn’t exist in the trials. Real world data let innovators build models for how effective the vaccines were likely to be against those variants. Similar models are being used to understand differential impact in subpopulations, the impact of multiple treatments, and the role of underlying health.
2. Uncovering rare opportunity
Real world data (RWD) has become a crucial engine for uncovering new evidence in rare disease. It starts with understanding the disease progression and overall impact on the patient over time. RWD charting that natural disease progression fuels important discovery and research design. But, increasingly, real world is standing in for both initial research and ongoing study over time. In some of the more than 7,000 rare diseases, there simply are not enough patients to support a typical trial. More importantly, many times the treatments are so essential to a patient population with significant unmet need that they simply cannot wait for traditional research.
3. Pro-patient approvals
In 2021, the FDA’s controversial approval of Biogen’s Aduhelm™ was welcomed by Alzheimer’s advocates and organizations across the US. During consideration of the drug’s surrogate endpoint and FDA precedent, those same advocates accused the agency of not being “pro patient.” For them, not giving them access to the drug meant not giving them access to hope. Even with the approval many questions remain about this and other drugs that were reviewed with surrogate endpoints instead of clinical ones. Real world data becomes a critical tool for the biopharma company and the agency to continue to build a picture of that drug over time, one focused on real world efficacy, safety and value. That use case appears even more often in rare diseases where complex, specialized rare gene and cell therapies that can alter DNA or RNA lead regulators to require five, 10 or even more years of data in the post-approval setting. Satisfying regulators, however, is just one step in bringing medicines to patients. Especially in rare diseases, manufacturers must also consider how payers will look at their evidence dossiers at a time of great change in the market-access environment. Thanks to a burst of research productivity, patients and doctors for the first time can choose among multiple therapies for rare conditions such as Duchenne muscular dystrophy and spinal muscular atrophy. In response, recent survey data5 show that payers will apply tools such as step therapies and formulary tiers to help manage costs. The emergence of treatment competition in rare conditions will change how payers look at a drug’s value proposition and continue to affect benefit design.
Manufacturers must anticipate these changes and upgrade their go-to-market procedures and philosophy, which may include accelerating the timelines on confirmatory trials so payers can be confident their budgets are spent on treatments that work.

Listen & Learn More About Navigating the Rare Disease Landscape
Flexible Decentralized Engagement
Every aspect of healthcare that relied on face-to-face engagement went from pilot to pandemic in 2020. Clinical trials were no different.
There had already been a heavy push in the industry to adopt technology-based applications to make clinical trials more efficient and to reduce the burden on patients, healthcare professions and sites, decrease delivery costs and increase quality.
Jump ahead: over half (54%) of trials now use an element of decentralization, including the ability to participate from home using remote monitoring and mobile nursing services.
That shift in trial experience and overall access to more flexible healthcare has permanently changed the expectations of clinical trial participants. Many will no longer be willing to take long drives to points of care for processes and monitoring that could be done at home or in the community. The concept that healthcare and waiting go hand-in-hand has been forever interrupted.
As we see the rapid uptick of patient choice in trial engagement around the world, the questions are rapidly turning from how to when.
Emerging Technologies and COVID-19
Remote Patient Monitoring is the Future of Standard Patient Care
as of 25 January 2021
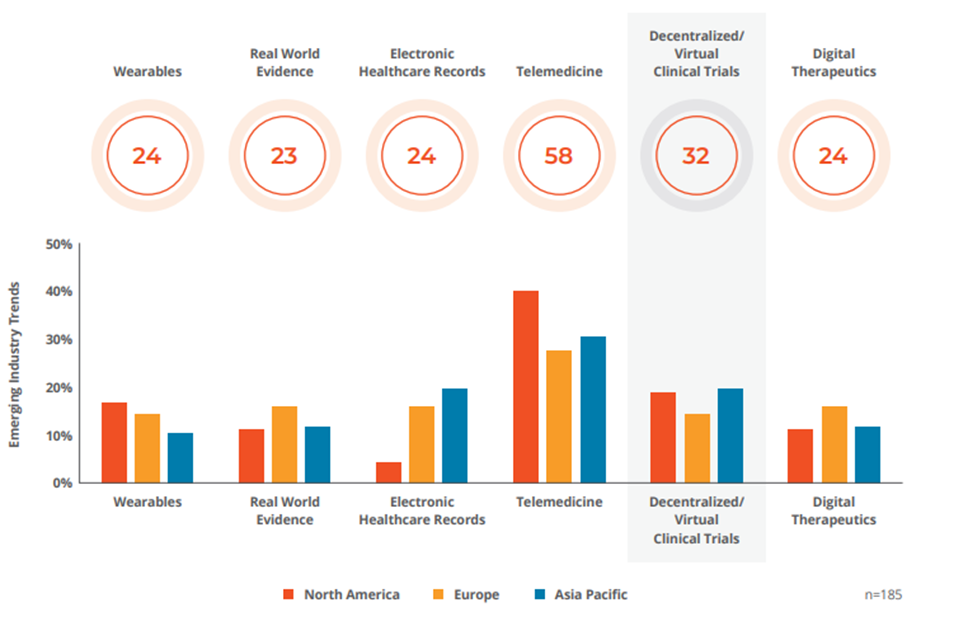
Q: Which life science industry trend will benefit the most from remote patient monitoring tools? Source: GlobalData’s Poll Conducted December 6, 2020 to January 12, 2021
The decentralized approach is essential for easing burden, expanding points of access and even promoting diversity in trials. But it’s not for every trial or every patient.
Even when patients are severely ill and a home-based protocol seems a natural fit, the approach may not match patients’ preferences—or the sponsor’s budget. These solutions must be applied with different stakeholder needs in mind. In the simplest terms, patients value optionality, trial sites need flexibility, and industry needs data integrity.
In 2022, look for more complex conversations about when to apply decentralized capabilities and how universally to do it. Some patients treasure in-person contact with their oncologists and medical staff. Or they just want to get out of the house and enjoy a routine—having lunch, grabbing coffee—that is woven into site visits. Others are balancing commitments from work to family that make travel to the site nearly impossible. They value convenience over all else. What happens when these cohorts are in the same trial?
Increasingly sponsors will consider the real world life experience of expected participants and balance that with what flexibility that can be built into the trial, giving participants as many choices as possible.
Need another example? Read, “It’s Time for Oncology Trials to Transition to Risk-Based Monitoring”
Dynamic Contracting
Underlying all these changes to the new clinical development model is partnership.
Sponsors are harmonizing solutions across multiple complementary services and technologies. The possibilities for acceleration require investments in time and labor as the operating model gets more complex.
Biopharma companies have responded by experimenting with different resourcing models and mixes of in-house delivery, staff augmentation, in-house delivery augmented by FSP (Functional Service Provider), in-house oversight as part of a FSO (Full-Service Outsourcing) model and more. This shift will likely gain momentum as organizations hedge against future supply chain disruptions.
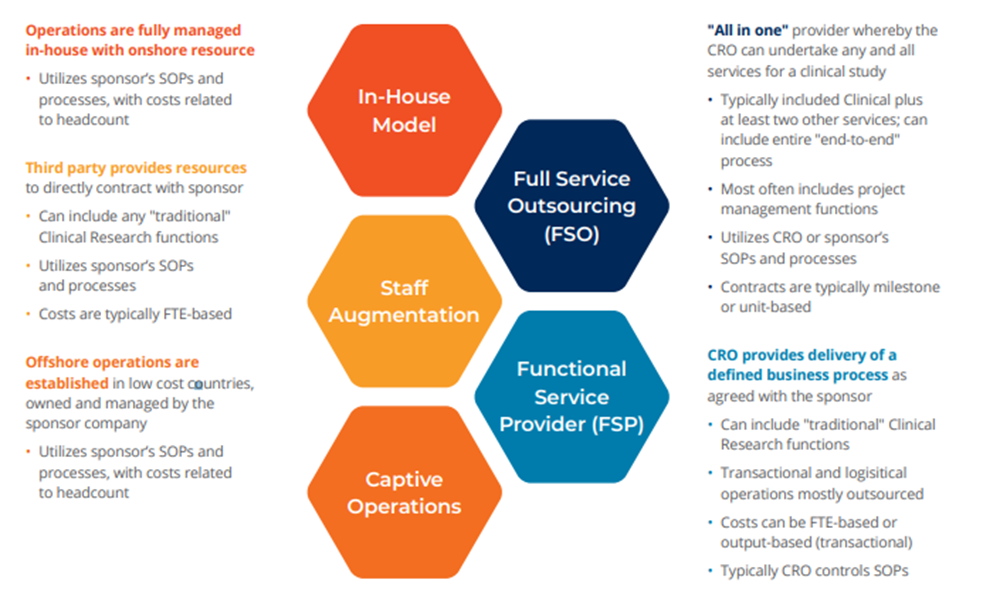
While no single model seems to out-perform the other industry-wide, there is evidence of a new trend toward building a hybrid that blends the advantages of each. The trend builds on the consolidation drive of recent years. With a limited pool of partners, biopharma leaders can ensure standardized training on shared systems and—importantly—shared ways of work. That creates flexibility to work across and within the models. Within an FSP relationship, for example, that hybrid model might enable a sponsor to dynamically select a specific component, trusting the quality and integration without additional contracting, cost variability, etc.
Get the printable PDF of Appropriate Acceleration in Clinical Development here.
Or, subscribe now to access the full 2022 Health Trends report and our monthly email to receive exclusive updates.